
Kashton (diagnosed with Alagille Syndrome), his mother Shauna, and Professor Dong
observing jagged mutant zebrafish.
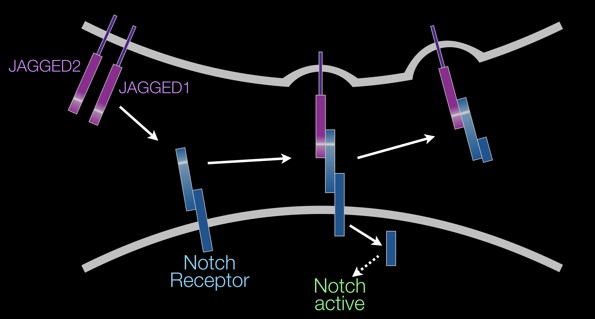
A simplified model of Jagged/Notch signaling showing Jagged ligands (purple) attached to the surface of
one cell (top; plasma membrane in gray) binds to a Notch receptor (blue) attached to the surface of an
adjacent cell (bottom) to activate Notch signaling through cleavage of the intracellular domain of the
receptor.
-
Alagille Syndrome (ALGS) is an autosomal dominant, developmental disorder with a prevalence estimated at I in
30,000 births. Although ALGS is characterized by developmental defects of multiples organ systems,
cardiovascular and hepatic pathologies (heart failure, aneurysm, and liver failure) are most life threatening.
-
ALGS is associated with heterozygous mutations primarily in JAGGED1, and less frequently in NOTCH2. Because
60-70% of ALGS cases are due to de novo (spontaneous) mutations, this disease will continue to persist.
-
Jagged/Notch signaling is required for the development of multiple tissues during embryogenesis. Binding of
the membrane-bound Jagged ligand from one cell to the membrane-bound Notch receptor of an adjacent cell allows
for direct cell-to-cell communication.
-
The current model for the neonatal liver duct paucity exhibited by ALGS patients, based on mouse model studies,
is that it results from a duct tube morphogenesis defect due to compromised JAGGED1 signaling from the liver
mesenchyme (port vein) cells. However, our recent studies using the vertebrate zebrafish model, support a different
mechanism – that biliary paucity is due a lineage specification defect due to insufficient Jagged signaling from
liver endoderm cells (Zhang/Gates et al., 2017, Nature Communications). It remains unclear whether either, or both,
mechanisms contributes to biliary paucity in ALGS patients.
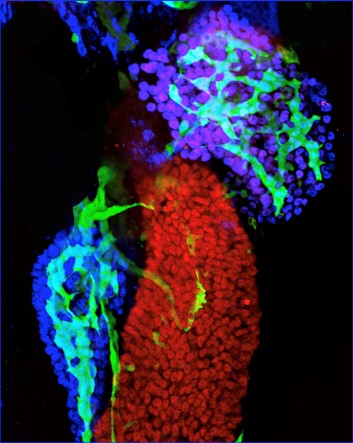
Notch active duct cells (green) in the pancreas (blue acinar cells) and liver (purple/blue hepatocytes)
can easily be labelled and imaged in the zebrafish. Zhang/Gates et al., 2017, Nature Communications
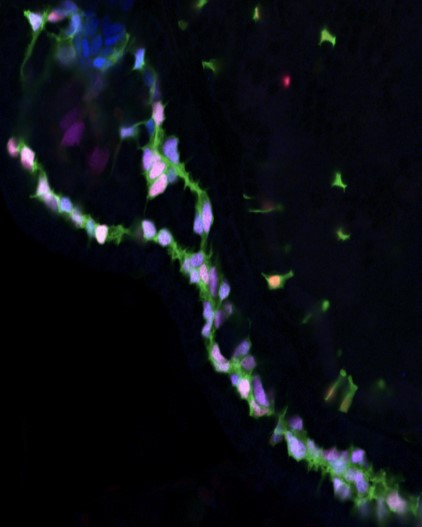
Duct cells (pink/lavender) all have and require Notch activity (green) for their lineage induction in the
zebrafish pancreas (above) and liver (not shown). Zhang/Gates et al., 2017, Nature Communications
Ongoing projects:
1. Genetic mechanism of JAGGED1 mutations in ALGS.
-
Understanding the mechanism of how heterozygous JAGGED1 mutations leads to ALGS pathologies is critical
for developing more informative animal models for ALSG. Cases of whole gene deletions of JAGGED1 indicate
a haploinsufficient mechanism. However, current heterozygous Jagged1 mutant mouse models exhibits ALGS
pathologies only in certain genetic backgrounds, limiting their utility for mechanistic studies and for testing
potential therapeutics. About 80% of JAGGED1 mutations associated with ALGS lead to a truncation and
loss of the transmembrane domain (ΔC-TM) which anchors the Jagged ligand to the cell surface. We aim to test
the hypothesis that most of the heterozygous JAGGED1 mutation alleles implicated in ALGS can exert a
dominant negative effect on Notch signaling (including Jagged2/Notch signaling), to consequently compromise biliary
lineage specification and other Notch regulated processes.
-
We are using the zebrafish vertebrate model to test whether these truncated forms can disrupt Notch signaling in a
dominant negative manner. Given our founding that Jagged2 plays a redundant role in liver duct lineage induction, we
will test whether Jagged1 transmembrane truncated ligands can disrupt both Jagged1/Notch and Jagged2/Notch signaling
to lead to more severe pathologies.
-
Because most of cases of ALGS are due to spontaneous mutations, it is imperative that we have a rapid approach to
assess the potential function consequences of these JAGGED1 mutations to inform genetic counseling efforts.
We are developing the zebrafish as a practical in vivo platform to functionally assess human JAGGED1 mutant
alleles.
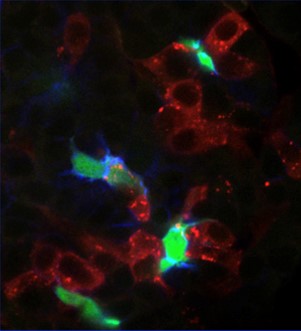
Our animal chimera studies demonstrate that normal liver cells (red) can rescue Jag/Notch signal and duct
lineage specification (green/blue) in neighboring cells of livers lacking Jagged expression. Zhang/Gates et al.,
2017, Nature Communications
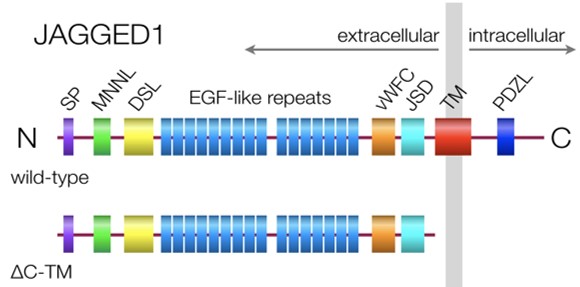
Diagram of a normal Jagged1 ligand (top, wild-type) attached to the surface of one cell (plasma membrane in gray)
with domains depicted. Most Jagged1 mutations in ALGS leads to a soluble form lacking the TM domain (bottom, ΔC-TM).
Domain abbreviations: SP, signal peptide; MNNL, Notch ligand N-terminal; DSL, Delta/Serrate/Lag2; vWFC, von Willebrand
factor type C; JSD, Jagged Serrate domain; TM, transmembrane; PDZL, PDZ ligand
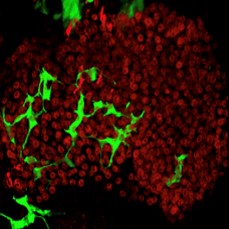
Liver (hepatocytes, red) with regenerated duct cells (green) following resumption of endogenous Jagged expression.
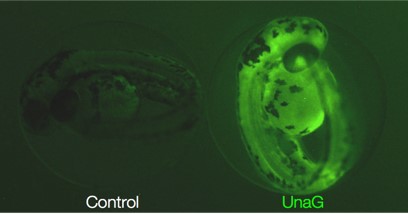
In vivo fluorescent reporter of bilirubin. Live fish expressing UnaG, which fluoresces when bound to bilirubin (right).
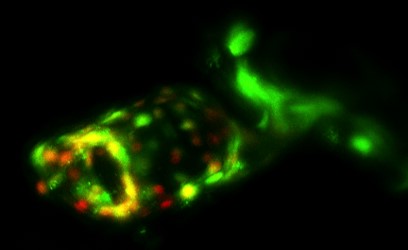
Endocardial cells (green) of the heart outflow tract are Notch active (red/orange/yellow) in zebrafish.
2. Reversing ALGS defects.
-
Developmental defects are generally thought to be permanent. However we are testing whether certain ALGS defects are an exception
and can be reversed. We are finding that complete developmental agenesis of liver biliary cells due to loss of Jag/Notch signaling
can be reversed if Jag/Notch signing is allowed to resume. We are investigating the origin of the regenerating duct cells, testing
our hypothesis that these regenerated biliary cells arise from the transdifferentiated hepatocytes. To examine whether these
regenerating biliary cells can restore liver duct structure and function, we are developing new genetic technologies to assess duct
network assembly and evaluate in vivo levels of bilirubin, a metabolite accumulated in patients with biliary paucity. Given the
potential reversibility of Jag/Notch defects and the amenability of the zebrafish vertebrate model for testing small molecules, we
will perform a chemical screen to identify drugs that will help resolve ALGS pathologies.
3. Live Flourescent reporter for bilirubin.
-
Liver duct paucity in ALGS patients lead to chronic cholestasis, a condition that causes elevated systemic bilirubin
levels. High bilirubin levels in ALGS patients are a predictor of long-term outcome of liver disease. The ability to
determine in real time whether a model animal has elevated bilirubin levels will be necessary to better understand the
dynamic functional effect of biliary paucity and regeneration. An in vivo indicator of bilirubin levels would
also yield unprecedented insight into possible levels differences among distinct tissues and organs. For example, it
would be insightful to assess levels of bilirubin in the brain, because bilirubin molecules can cross the blood brain
barrier, accumulate, and become neurotoxic. By expressing a protein (UnaG) that fluoresces upon bilirubin binding, we
are assessing bilirubin levels in live zebrafish mutant models of ALGS. Particularly we are investigating how the
dynamic state of loss or regenerating biliary duct cells can affect bilirubin levels.
4. Mechanism of Jagged function in liver, heart, and vasculature.
-
With our discovery of new mechanisms for Jag/Notch signaling in liver duct cell development, we are continuing to leverage
the unique genetic and imaging techniques available with the zebrafish vertebrate model system to rigorously explore the role
of Jag/Notch signaling in the development and maintenance of other tissues affected in ALGS, particularly the heart and vascular
system. Uncovering different new mechanisms of Jag/Notch function in these tissues will reveal new therapeutic potential for
ALGS.
Under construction. See our video for a preview about our exciting
breakthrough on directly reprogramming cell lineage, in vivo. Check back later for details and updates.
-
The broad classification of disease subtypes often masks their mechanistic heterogeneity as well as their potential similarities.
For example, non-autoimmune diabetics are typically classified as early or late onset diabetes, or polygenic or monogenic diabetes,
i.e., Type 2 Diabetes (T2D) versus Maturity Onset Diabetes of the Young (MODY). Based on the genes implicated in T2D by genome wide
association studies (GWAS) and in monogenic diabetes, a variety of different genetic mechanisms may contribute to this disease,
supporting the idea that T2D is comprised of multiple etiological classes. By recognizing and understanding a patient’s disease
subtypes based on genetic mechanism, we will be better able to design more targeted drug screens to discover new therapeutics that
are more personalized.
-
We are currently focusing on HNF1B and HNF1A associated diabetes. HNF1B and HNF1A are two of few genes implicated in both T2D and
monogenic diabetes. Our lab has developed the only existing vertebrate genetic animal model that reliably mimics the pancreas defects
associated with HNF1B diabetes (MODY5). Furthermore, we have generated and are characterizing a new mutant model for HNF1A diabetes
(MODY3). We are currently utilizing these zebrafish mutant diabetes models to investigate their role in normoglycemia and uncover
genetic interactions with other genes implicated by T2D GWAS. Together to with studying the function other genes implicated in T2D,
we aim to reveal distinct mechanisms of diabetes based on the genes affected in individual patients – to ultimately allow for
personalized diabetes therapeutics.
-
Personalized Medicine – As more people gain access to their individual genetic data, more questions arise regarding anomalies uncovered
from the analysis of their genomic sequence. While certain anomalies can be accessed based on published research or by a predictive
bioinformatics approach, others will require more definitive functional studies to better understand the potential affects of their
mutation on gene function and health. Our lab’s expertise in liver and pancreas biology allows us to investigate genetic anomalies
associated with diseases related with these organs. With animal models and technologies that we are developing in the lab, we can rapidly
examine the expression and function of the genes effected, modeling the effect of the mutation in an in vivo vertebrate system. Our goal
is to rapidly access the functional consequences of human disease mutations and determine their pathological mechanisms.
|